Potential Mutation Errors in Gene Bounds: Implications for the Evolutionary Status of A/T and CpG
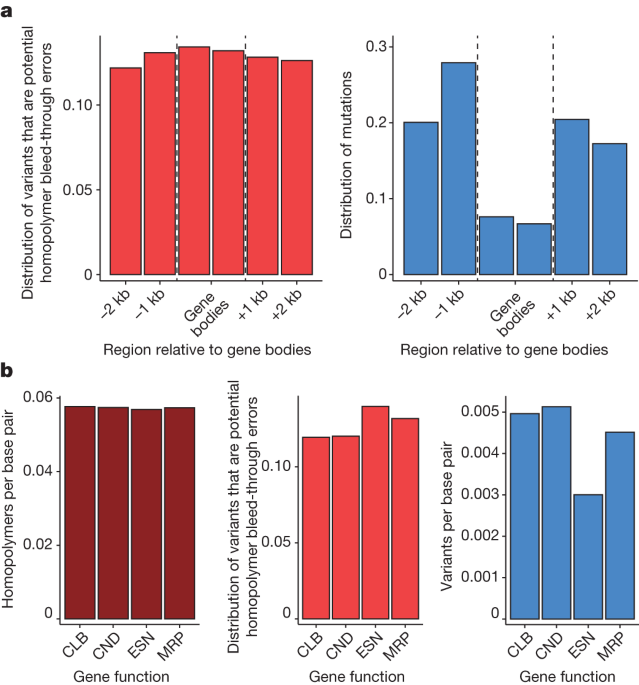
Wang and colleagues report that the proportion of potential bleed through errors in our data is higher than expected, which should lead to underestimates of Gene body Mutation rates. We can confirm that the proportion of potential bleed-through errors is not lower in the same group of genes as it is in the other group. Similarly, the proportion of potential homopolymer bleed-through errors is not reduced in essential genes (Fig. 1b). The hypothesis of Wang and colleagues is not in agreement with the distribution of potential bleed-through errors. By contrast, the observed pattern is expected if gene bodies and essential genes experienced a reduction in true mutation rates, as noise introduced by sequencing errors should have a proportionally larger effect on regions with truly low mutation rates.
For Monroe’s somatic mutations (recalled from Weng’s vcf data) unassociated with homomeric runs (46% of their mutations), most are clustered (2 mutations within 10 base pairs of each, 27.5% of all mutations) or unexpectedly common (>10 samples, 8.7% of all mutations), indicative of mis-mapping issues. 2.5% of the Weng HQ data is clustered. Many of Monroe’s putative mutations are associated with more than one error: about 34% are associated with A/T homomeric runs and in a tight cluster. Mis- mapping, a trait of centromeres, may explain why 49.9% of LQ is centromeric. 1e) compared with 27.9% in Weng HQ.
This neighbour base matching affecting both A and T in Monroe’s data is an expected bleed artefact with no biological basis. By contrast, we expect CpG>TpG mutations to be common given well-described methylated CpG hyperinstability15. This is the case in Weng’s data but less so in Monroe’s.
Monroe claims that there are lower rates of decay at more important sites but the rate is not uniform. In some part this is because transposable elements (TEs) have high mutation rates and TEs are rare in gene bodies. C stability should be a result of the suppression of the cytosine genes in Arabidopsis. In the Weng data, 65% of mutations in TE are CG, CHH or CHG versus 51% in intergenic non-TE (for example, 5.2:1 ratio of CpG>TpG per CG, TE to intergenic non-TE). In (robust) germline data there is a higher mutation rate in TEs than elsewhere, including the best comparator, non-TE intergenic sequence: TE versus non-TE intergenic sequence, mean ratio per dinucleotide = 3.93 (paired t-test on normalized dinucleotide rates, P < 3 × 10−7, df = 95). This TE enrichment largely explains why in germline data mutation rates are higher 5′ of TSS and 3′ of TTS, TEs being enriched outside transcribed domains (Fig. 1a).
Wang and colleagues1 assume that homopolymer bleed-through errors affect sequences up to five nucleotides away from homopolymers, although these errors occur on modern Illumina platforms at positions immediately adjacent to a run of identical bases7. Moreover, their simulation of sequencing errors apparently assumes that 100% of sequencing errors occur as a product of homopolymer bleed-through. The estimates of the result ofHomopolymer bleed-through7 are only 0.7 to 5.2%. Across all data, 12.0% of single-nucleotide variant calls could be the potential source of bleed-through errors, but on their own cannot explain the 50% reduction in genes we observed.
We conclude that the reported relationships between epigenomic features and mutation rates2 are well supported mechanistically (Fig. 2e). It’s difficult to know the accuracy of individual calls in the huge set of wildly different variants because of issues and inherent uncertainties that have to do with the very large set. However, the proposal that the observed patterns result only from sequencing errors is inconsistent with multiple lines of evidence from the original study, independent analyses and emerging parallel work.