Cellular ownership of defective mitochondria can cause cancers to become ill-equipped and death-successful: the case of TILs
Cancer cells can poison attacking immune cells by filling them with defective mitochondria ― dampening the body’s defensive forces and helping the tumour to evade eradication1.
J.R.B. is a member of the Scientific Advisory Board of LUCA Science, Inc., receives research support from LUCA Science and Edgewise Therapeutic, has consulted for Columbus Instruments within the past year, and is receiving royalties from Decibio.
Jonathon Brestoff works at the Washington University School of Medicine and he says this changes our view of cellular ownership of mitochondria.
“My first thought was that this sounds crazy, like science fiction. But they seem to have the data for it,” says Holden Maecker, an immunologist at Stanford University in California, who was not involved in the research. This is a completely new biology that we were not looking at.
In cellular models the team showed thatinted TILs were less able to divide and commit suicide. T cell exhaustion was shown in mice with cancer, when TILs that had infused alien mitochondria.
Multivariate in vitro anti-PD-1 therapy in a tumour microenvironment: a statistical analysis and a t-test
All in vitro experiments were biologically repeated independently three to four times and produced consistent results. Consistent results were produced from all the in vivo mouse experiments conducted with at least six mice per group.
The patient characteristics were compared using Fisher tests. The variables were compared using a t-test and one-way ANOVA. Two-way ANOVA was used to compare the Tumour volume curves. The Bonferroni correction was used for multiple testing. Progression-free survival and overall survival were defined as the time intervals from the initiation of anti-PD-1 monoclonal antibody therapy until the first observation of disease progression or death from any cause, and until death from any cause, respectively. The survival curves were analysed using the Kaplan–Meier method. The tests weretailed with a significance level. GraphPad software was used to perform statistical analyses. The means and error are shown.
Source: Immune evasion through mitochondrial transfer in the tumour microenvironment
Controlled cell coculture with Mito Green cells in water and injected with FCCP-based anti-rabbit antibody in OT-1 mouse models
The cells were labeled Mito Green. Twenty-four hours later, TIL04#9 cells were cocultured with MEL04-MitoDsRed cells for 3 days with or without 10 mM NAC. Subsequently, the cells were stained with an LC3B-specific polyclonal antibody (Proteintech, Rosemont, 18725-1-AP) followed by an APC-conjugated secondary antibody (goat anti-rabbit IgG, Abcam, ab130805), and observed under a confocal laser microscope or analysed by flow cytometry. We used 10 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (Selleck Biotech) as a positive control.
LLC/P29-MitoDsRed cells (1 × 105) or LLC/A11-MitoDsRed cells (2 × 105) were subcutaneously inoculated into OT-1 mice. The effector cells were cocultured with Calcein-AM and sorted from the TILs after 42 days. The cells were used as the controls. After 3 h of the sample being exposed to the water, a set of 490/535 nm on an arovo X3 Multilabel reader was used to determine the status of the sample.
All mice were maintained under specific pathogen-free conditions at the animal facility of the Institute of Biophysics (Chiba Cancer Center Research Institute and Okayama University). All mouse experiments were approved by the Animal Committee for Animal Experimentation of Chiba Cancer Center and Okayama University, and met the US Public Health Service Policy on Humane Care and Use of Laboratory Animals. When the maximum tumour diameter exceeded 20 mm, mice were killed as a humane end point. The experimental schematic is summarized in supplementary fig. 3.
Female C57BL/6J mice (6–8 weeks old) were purchased from SLC Japan. C57BL/6J- The National bioResource Project of the Japan Ministry of Education, Culture, Sports, Science and Technology/Agency for Medical Research and Development provided PrkDCscid>/Rbrc mice. The Jackson Laboratory purchased OT-1, Cd4cre, and Tfamfloxed mice. The Rat anti-mousePD-1 and anti-mouseCD8 were obtained from Bio X Cell. The control rat IgG2a monoclonal antibody (RTK2758) was obtained from BioLegend.
Source: Immune evasion through mitochondrial transfer in the tumour microenvironment
IncuCyte ZOOM System for the evaluation of cellular proliferation, cell ensoucination and oxygen consumption rates. IV. Application to TIL09#9 cells
Twenty-four hours after the passage of cells on 96 well plates, in cellular proliferation was evaluated using the IncuCyte ZOOM System.
Annexin V was used to evaluate apoptosis. It’s possible to fix ebioscience. Dye eFluor (Thermo Fisher Scientific) for live/dead cell staining. According to the manufacturer’s instructions, each cell was incubated with Annexin V and eFluor for 15 min at room temperature and then analysed by flow cytometry. Cellular proliferation was assessed on the basis of the dilution of cells labelled with carboxyfluorescein succinimidyl ester (CFSE) using a CFSE Cell Proliferation kit (Thermo Fisher Scientific) and flow cytometry. For 20 min at 37C in 5% CO2, cells were washed 3 times and then incubated for 3 days with 10 M CFSE and a RPMI medium.
According to the instructions of the manufacturer, a Cellular Senescence Detection kit was used. The cells were put to use in a single day and then put to use for 30 minutes and analysed by flow cytometry.
Metabolic analyses were performed using a flux analyser (Seahorse XF HS mini, Agilent Technologies), according to the manufacturer’s instructions. In a single step, 0.8 105 cells were seeded in supplemented Seahorse RPM I medium containing 1 mM pyruvate, 2 mM glutamine and 10 mMglucose, in poly-D-lysine-coated XFp miniplates. The plate was then equilibrated at 37 °C in an incubator without CO2 for 40 min. The oxygen consumption rate was evaluated with injections of FCCP and rotenone. The rate was evaluated by the injections of the three hormones, as well as 2-deoxy-glucose. The rate of production of the molecule was evaluated by sequential injections of the two drugs. All chemicals were purchased from Agilent Technologies. All data were normalized to the cell number.
TIL09#9 cells were labeled with Mito Green and cocultured with MEL03-MitoDsRed for 3 days. TIL04.#9 cells were stained with either an anti-USP 30 monoclonal antibody or an anti-USP 30 polyclonal antibodies, both of which have the same binding site.
The Mitochondria Isolation kit was used to get rid of the MEL03, MEL05, and MEL06 cells from the cultured cells. The cells were subsequently cultured for 24 h after centrifugation, with 20,000g for 15 min. Four times a week this procedure was repeated. Adding the isolated EVs to Jurkat/Rho0 cells using the EV-Entry system was done in 15 min at 4C. These EV-transferred Jurkat/Rho0 cells were cultured for 6 weeks, and this procedure was repeated every 5–7 days.
MEL02-MitoDsRed cells or MEL03-MItoDs Red cells were used to coculture the naive T cells from the healthy donors. The central memory fraction and KLRG1 expression level were analysed by flow cytometry. Each CD8+ T cell fraction (naive, CCR7highCD45RAhigh; central memory, CCR7highCD45RAlow; effector memory, CCR7lowCD45RAlow; terminally differentiated effector memory, CCR7lowCD45RAhigh) sorted from PBLs of healthy donors was also cocultured with MEL02-MitoDsRed cells or MEL04-MitoDsRed cells for 4 days in the presence of IL-2 (300 IU ml–1) alone, and apoptosis was analysed by flow cytometry.
The MC-38 cell line (mouse colon adenocarcinoma) was purchased from Kerafast, and the B16F10 (mouse melanoma), MCF7 (human breast cancer), MDA-MB-231 (human breast cancer) and Jurkat (human T cell leukaemia) cell lines were purchased from the American Type Culture Collection (ATCC). The LLC/P29 and LLC/A11 (mouse lung carcinoma) cell lines were established from mouse Lewis Lung carcinomas as previously described17,64,65. The cells were kept in a modified Eagle’s medium, which contained 10% FBS. mtDNA-deficient Jurkat (Jurkat/Rho0) cells were generated by culturing Jurkat cells in the presence of 200 ng ml–1 ethidium bromide for 6 weeks and then maintained in RPMI1640 medium containing 10% FBS, 1% PS, 100 μg ml–1 sodium pyruvate and 50 μg ml–1 uridine.
MitoCheck Activity Assay kits (complexes I, II and III, and IV) were purchased from Cayman Chemical. Mitochondrial protein isolation from cell lines was performed using a Mitochondria Isolation kit for cultured cells (Thermo Fisher Scientific) according to the manufacturer’s instructions. Following the instructions given by the manufacturer, we used activity buffer with the isolated MIP in place of the supplied mitochondria. Reactions were conducted at 25 °C using a FlexStation 3 microplate reader (Molecular Devices), with readings taken every 30 s for 15 min at the Central Research Laboratory of the Okayama University Medical School.
We used a DCFDA/H2DCFDA Cellular ROS Assay kit (Abcam) to detect ROS. In 30 min, the cells were put into 50 M DCFDA solution and then into 5% CO2 at 37 C. Next, we extracted and purified EVs from the cells and analysed them by flow cytometry with a PS Capture Exosome Flow Cytometry kit (Wako) to create EV-conjugated beads.
Source: Immune evasion through mitochondrial transfer in the tumour microenvironment
Holographic microscopy of USPS data from TCGA: Coculture and labeling of red and white cells with pBABE-puro antigens
We obtained RNA-sequencing expression data from The Cancer Genome Atlas (TCGA) in the Genomic Data Commons data portal of patients with melanoma from the USCS Xena database (https://xenabrowser.net). USP30, USP33 and USP35 expression data in tumour tissues were used.
For time-lapse imaging, 2 × 105 MEL04-MitoDsRed cells were seeded onto a 35-mm glass-bottom culture dish 24 h before coculture and allowed to adhere. The following day, coculture with 1 × 106 TIL04#9 cells labelled with MitoTracker Green was initiated. Twenty-four hours later, we began capturing images every 30 min using a digital holographic microscope (3D Cell Explorer CX-A, Nanolive). The images were analysed using Fiji software (https://imagej.net/software/fiji).
TIL09 and MEL09 cells were labeled with MitoDsRed and MitoTracker Green. After 24 h, cells of the red variety were cocultured in a 35-mm glass bottom dish with cells of the white variety under a confocal laser microscope. TIL04#9 cells were labelled with a BV421-conjugated monoclonal antibody specific for CD45 (clone HI100, BioLegend).
Addgene purchased pBABE-puro (1764)67 and pVL-MitoDsRed. The manufacturer gave instructions for the cloned TfR-OVA to be used as a pBABE-puro vector. The resulting pBABE-puro-TfR-OVA vector was transfected with a pVSV-G vector (Takara Bio) into packaging cells using Lipofectamine 3000 reagent (Thermo Fisher Scientific). The pVL-MitoDsRed was transfected with a variety of plasmids. After 48 h, the supernatants were transduced into the cell lines MEL03, MEL06, MELc03 and B16F10, among others. The cells were transfected using the MELc03-mitoDsRed, MCF7-MItoDsRed, and MEL02-mitoDsRed. There were also generated cell lines.
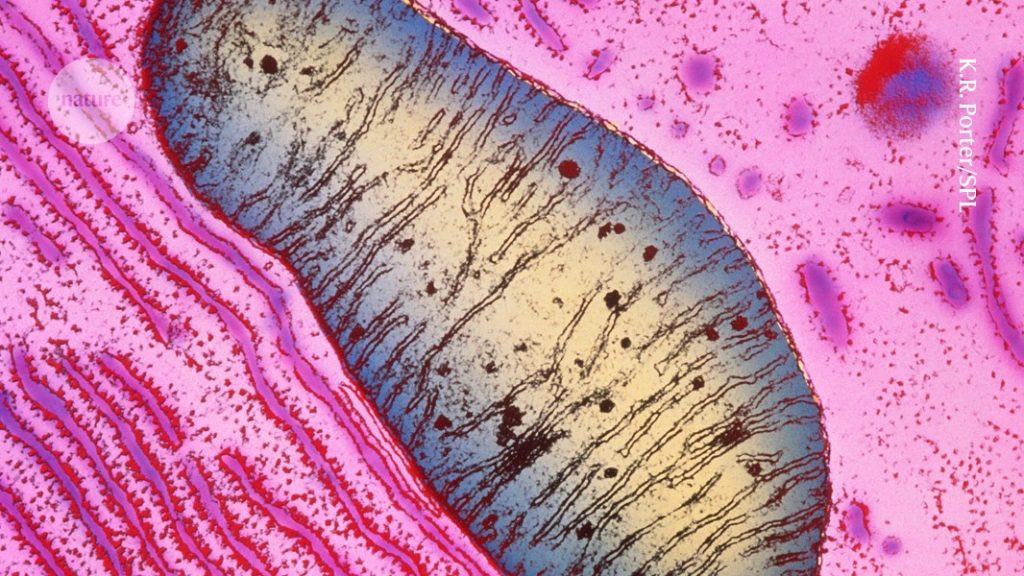
For pre-fixation, cell specimens were immersed in 0.1 M PBS, pH 7.4, containing 2% glutaraldehyde and 2% paraformaldehyde for 16–18 h. Post-fixation was performed with 2% osmium tetroxide for 1.5 h. The specimen were dehydrated and embedded in the Spurr Resin after washing with PBS. The sections were stained with uranyl Acetate and lead citrate after being prepared using an ultramicrotome. The specimen was observed using a transmission electron microscope. We counted and quantified the number of cristae per mitochondrion.
Source: Immune evasion through mitochondrial transfer in the tumour microenvironment
Immunity evasion through mitochondrial transfer in the microenvironment of a human-tumor cell incubated in RPMI1640 medium
All cell lines were used after confirming that they were mycoplasma-free, which was assessed using a PCR Mycoplasma Detection kit (Takara) according to the manufacturer’s instructions.
To establish cancer cell lines, 1 × 107 digested tumour cells were cultured in RPMI1640 medium containing 10% FBS (Cytiva), 1% penicillin–streptomycin (PS) and 1% amphotericin B (Thermo Fisher Scientific). When Tumour cells were free of fibroblasts, they’d use them when they were able to get beyond the tenth passage. To establish and expand cultured TILs, tumour digests were incubated in RPMI1640 medium supplemented with 10% human AB serum, 1% PS and recombinant human interleukin-2 (rhIL-2: 6,000 IU ml–1, PeproTech) in a humidified 37 °C incubator with 5% CO2. A half of the medium was taken from the wells and replaced with fresh medium every few days.
Source: Immune evasion through mitochondrial transfer in the tumour microenvironment
Using the frequency frequency per position metric to evaluate the existence of real variants in normal and tumour samples for cell line selection and TILs
We used the Z score from the variant allallefrequency per position as a metric to evaluate possible variations after pooling the results from all samples. Alterations with z scores > 3, VAF > 0.2 and read depths exceeding 100 were selected to minimize false positives from sequencing errors. In addition to the assumption that there would not be high VAFs due to the presence of a significant fraction of non-cancerous cells in the specimen, the variants with VAF > 0.85 were excluded from final reporting. Haplogrep (v.2.4.0; Kulcyznski classification mode with 17_FU1 phylotree, 10 top hits, extended final output and heteroplasmy levels set to 0.85)63 was used to characterize potential haplogroups of individual cases and to identify possible unlabelled SNPs from our frequency-based selection criteria. There were two types of variant: hotspot and local private variant, both of which were labeled as polymorphicVariants were deemed to be polymorphic when they were labelled as hotspot andlocal private variant. In order to assess performance we compared the existence of true and false variants, using samples from both normal and tumours. Using these criteria, true variants were called with a false-positive rate of 0% and a false-negative rate of 12.2%.
The protocol for this study was approved by the appropriate institutional review boards and ethics committees of Yamanashi University Hospital, Chiba University Hospital, Shinshu University Hospital, Okayama University Hospital, Kindai University Hospital and Saitama Medical University International Medical Center. This study was conducted in accordance with the principles of the Declaration of Helsinki.
One of the patients with melanoma had a sample used to establish TILs and match cancer cell lines, along with one of the others with breast and skin cancer. The participants had undergone surgery at Yamanashi University Hospital. The tumor tissues were processed before. In brief, surgically resected samples were enzymatically digested with 0.1% collagenase, 0.01% hyaluronidase and 30 U ml–1 deoxyribonuclease (Sigma-Aldrich) in RPMI1640 (Thermo Fisher Scientific) at room temperature. The tumour cells were subjected to separation before use. Blood mononuclear cells could be obtained by using Ficoll–Uropoline density-gradient centrifugation. All participants gave their written consent.